Development of force fields used in molecular dynamics calculation
Molecular Dynamics (MD) are one of the molecular modeling and simulation method and nowadays a widely used. In this article, we will review the classification and recent development of force fields used in MD calculations.
A force field is an empirical expression for the forces and energies required for MD calculations, and the parameters included in this expression are called force fields parameters. Various force fields have been developed since the 1970s. This article will introduce the classification of force fields, mainly targeting organic systems, and will also touch on recent developments such as reactive force fields and machine learning force fields.
1. Molecular dynamics and forces acting on atoms
First, we show what is doing in MD calculations.
In MD, the forces acting on a particle at a given time are calculated and its motion is solved based on Newton's equations. This can be repeated sequentially and analyzed for a long period of time. The time variation of the particle position x can be expressed by the Taylor expansion as follows;
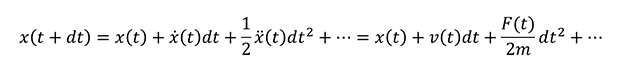
In this equation, x with . (dot) is the first-order derivative of position x with time, .. (two-dot) x is the second-order derivative, corresponding to the velocity v and force F respectively.
In actual MD calculations, Verlet's method or others are commonly used, although the derivation is omitted, and the time integral is as follows.
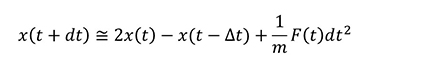
This means that the force F is required for MD calculations.
Forces and energies can be calculated using ab initio calculation methods (e.g. density functional theory, DFT) and such methods are actually used. This is called ab initio MD calculations (AIMD) and is a useful method for MD calculations in systems where no force field parameters available.
However, it is very expensive, and in usual (classical) MD calculations, the forces were calculated using empirical formulae.
2. Classification of force field used in molecular dynamics
Classical force fields are classified as Class I, II and III.
Class I force fields consist of the potential energy U is bond, angle, torsion and LJ and electrostatic (ES) interaction, as shown below. DREIDING, AMBER, GAFF and OPLS are examples of such force fields.
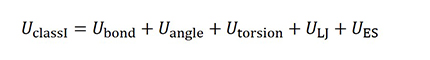
Class II force fields include bond-bond and bond-angle coupling terms in addition to the Class I potential energy. PCFF is an example of such force field.

Class III force fields have been proposed in addition to Class II force fields, including those that further incorporate charge polarization effects, i.e. the effect of a fluctuating charge distribution in time.
(In Class I and II, the charge is essentially a fixed point charge on an atom.)
Although these force fields can deal with molecules in a generic way, there are some molecules for which specially tuned models have been proposed.
For example, many tuned models have been proposed for water, with the SPC, SPC/E, SPC-FW, TIP3P, TIP4P and OPC3 known as typical examples.
Although a water atom consists of three atoms, some water models have three interaction points and others have more than four, with TIP4P being a four-point model. Also, in most models, except SPC-FW, water molecules are treated as rigid bodies, so MD calculations are performed under appropriate constraints, such as SHAKE and RATTLE methods.
*TIP3P and SPC-FW parameters can be used in J-OCTA/COGNAC modeler.
The SPC/E, TIP3P and OPC3 models can be used in the GENESIS modeler.
There are differences in the physical properties calculated for these models.
For more information, see the following pages.
https://water.lsbu.ac.uk/water/water_models.html
This form of force field is mainly used for organic molecules, whereas different types of force fields, EAM, Tersoff, etc., are used for inorganic materials. In some cases, such as at organic-inorganic interfaces, there are no available force field parameters, and there are attempts at DFT-MD linkages to determine the non-bond interaction parameters based on DFT calculations. [1]
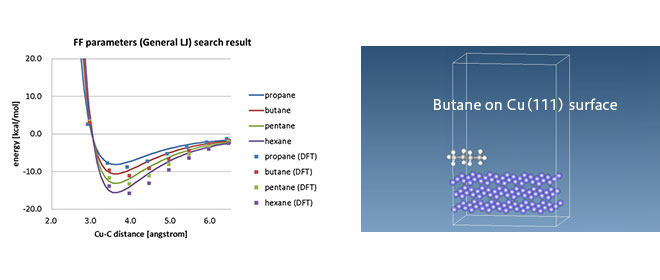
*J-OCTA provides an LJ parameter determination function using SIESTA as DFT software, allowing MD calculations of organic-inorganic interfaces.
[Case study] DFT-MD study for interfacial systems
The force fields described above basically deal with the stable state of the molecule, but since the 2000s, reactive force fields have been proposed such as ReaxFF[2], which can also include reaction processes.
There are two main types of MD calculations involving reactions: in the case of the reaction process is to be followed in detail and in the case of the structure after the reaction is of interest. ReaxFF is used for the former, where the reaction process is followed in detail.
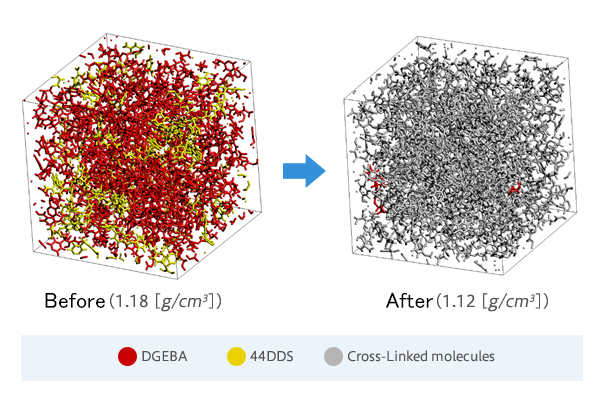
* There is the case study of J-OCTA/VSOP, which focus on the structure after the reaction of thermoset resin.
[Case study] Cross-Linking of Epoxy Resin by the Monte Carlo method using activation energy
Recently, machine learning-based force fields (ML-FFs) have been proposed.
While ML force fields offer a level of accuracy close to that of AIMD, the computational cost is greater than that of classical force fields. Although it has been applied mainly to inorganic materials first, it has also been applied to organic systems and is expected to be developed in the future. [3-5]
The types and overview of the force fields introduced in this article are summarized in Table 1.
In 2023, MD with classical force fields using HPC environments and cluster machines is becoming the standard for calculations of 100 nanoseconds or longer. ML force fields have a larger computational cost than classical force fields, so the application tasks should be chosen carefully, as it is difficult to calculate for as long a time as classical MD. The calculation speeds listed in Table 1 are approximate. the scaling is different for AIMD and the others, but it should hopefully serve as a guide when considering them. [6]
Table1. Types of force fields used in Molecular Dynamics
| Force field | Contribution to the energy | Force field sets | Calculation speed |
|---|---|---|---|
| Class I | Bonding and non-bonding | AMBER, GAFF, OPLS, others | 1 |
| Class II | Coupling terms are added to Class I | UFF, PCFF, others | : |
| Class III | Polarization effects etc. are added to Class I/II | AMOEBA, others | : |
| Reactive FFs | See reference [2] | ReaxFF, others | : |
| ML-FFs | See reference[3-5] | DeepMD, others | 0.01 |
| Ab initio | Based on DFT etc. | Not required | < 0.0001 |
- Reference
- [1] K. Johnston and V. Harmandaris, J. Phys. Chem. C, 115 14707 (2011)
- [2] A.C.T. van Duin, et al., J. Phys. Chem. A, 105, 9396 (2001)
- [3] V. L. Deringer, et al., Chem. Rev., 121, 10073 (2021)
- [4] E. Kocer, T. W. Ko and J. Behler, Annu. Rev. Phys. Chem., 73, 163 (2022)
- [5] T. Wen et al., Mater. Futures, 1, 022601 (2022)
- [6] L. Zhang, et al., Phys Rev. Lett., 120, 143001 (2018)