- Quantum Chemistry / DFT
- Interface / Phase Separation / Particle Dispersion
- Materials Science
[Analysis Example] Surface Reconstruction
Calculation of the surface structure of Si
A solid surface is energetically less favorable than the bulk state because the atoms that make up the surface lose their bonds with surrounding atoms. The magnitude of the energy loss is known as the surface energy, which has different values depending on the surface orientation (ref:[Case Studies] Surface Energy of Pigments).The electronic state near the surface is different from that in the bulk, and the face spacing near the surface of metals and other materials is known to deviate from that inside the crystal. In covalent solids, it is known that not only one-dimensional structural relaxation but also two-dimensional relaxation occurs in the plane.
Here, the relaxation of the surface structure of Si(001) is investigated by ab initio MD calculations.MD calculations for the Si(001) surface were performed using the MD calculation function of the SIESTA modeler. We used PBE as the functional and SZP as the basis function. The time step was 2fs and the total calculation was 1ps. In this calculation, the atoms in three layers near the surface were excluded and constrained. Figure 1 plots the potential energy during the MD calculation, and the snapshot at 0.8 ps shows the formation of the dimer structure. From the change in potential energy, we can see that it goes beyond some energy valley structures, and it is possible that the dimer structure is not reached by optimization calculations.
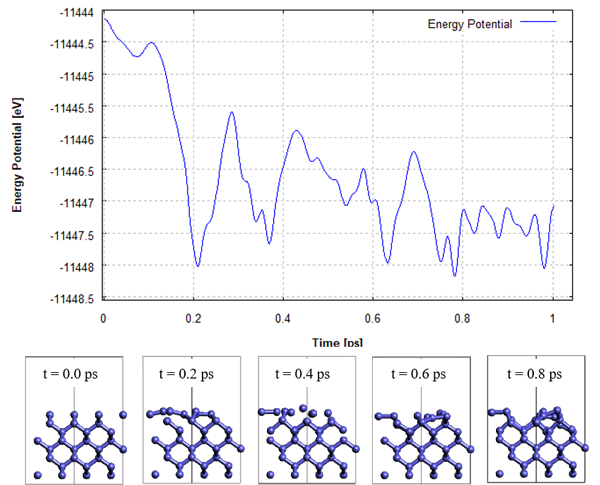 Fig.1 ab initio MD calculation of surface reconstruction of Si(001)
Fig.1 ab initio MD calculation of surface reconstruction of Si(001)
Calculation of the surface structure of SiO2
A similar example of a calculation is the creation of a Q4 surface of silica using ab initio MD. In a Q4 surface, all the surface oxygen is bonded to Si atoms, which is known as a hydrophobic surface. This surface has a characteristic ring structure. The MD calculation uses the structure of quartz as the initial structure. The desired structure was obtained after 200 steps of calculation with a time step of 2fs.
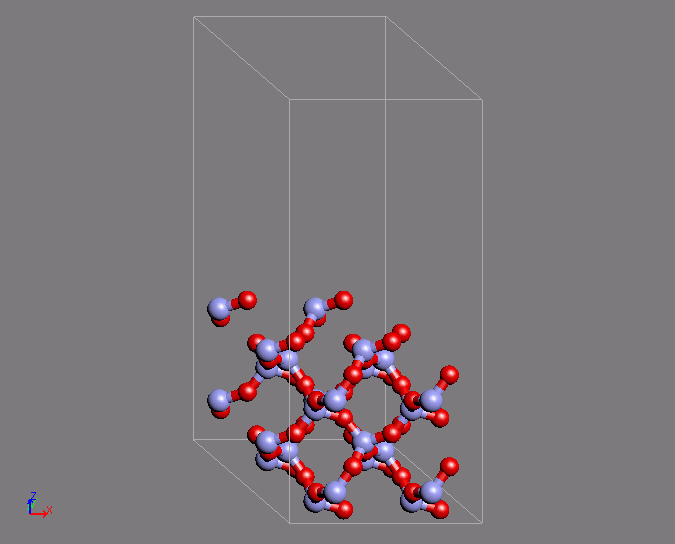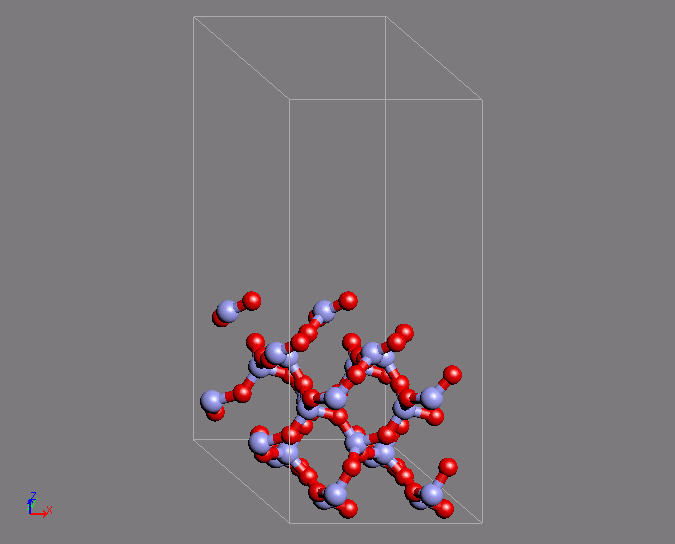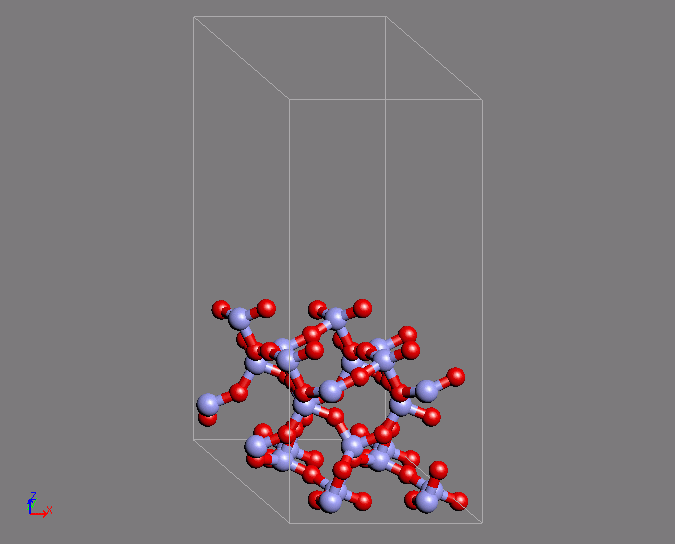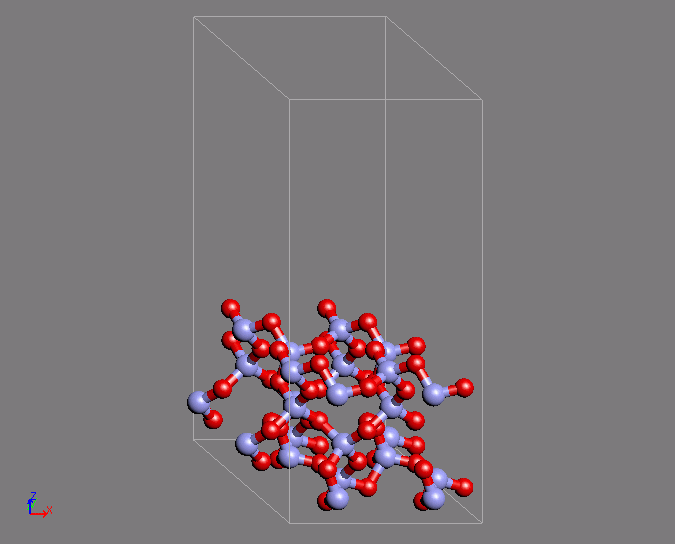
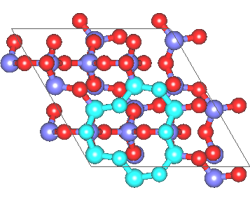
Fig.2 Silica Q4 surface structure by using ab initio MD calculations
- (left) Initial structure
- (right) Characteristic ring structure on the surface