Functions
From molecular characteristics to material properties
- - First-Principles calculation (engine name: SIESTA)
- - Molecular dynamics simulation (engine name: COGNAC, VSOP, GENESIS)
- - Interface/phase separation simulation (engine name: SUSHI, COGNAC-DPD)
- - Rheology simulation (engine name: VSOP-DPD, PASTA, NAPLES)
- - Multi-phase material, RVE, and Process simulation (engine name : MUFFIN, VSOP-PS)
The modeling tools provided in J-OCTA for each engine are introduced here. Interfaces with other software, the QSPR function for the simplified calculation of polymer properties, and inter-scaling coupling functions - including zooming and reverse mapping - are available.
Review Papers
- Latest Functions of J OCTA, Multiscale Simulation Software for Soft Materials
Journal of the Imaging Society of Japan,Vol.57, No.6, P.700 705 (2018) - Multiscale Simulation Technology and Software for Soft Matter
Journal of the Imaging Society of Japan,Vol.54, No.6, P.578 586 (2015)
-
 Crystal Structure and Molecular Modeling
Crystal Structure and Molecular Modeling -
 First-Principles Calculation (SIESTA)
First-Principles Calculation (SIESTA) -
 Molecular dynamics simulation (COGNAC, VSOP, GENESIS)
Molecular dynamics simulation (COGNAC, VSOP, GENESIS) -
 Interface, phase separation simulation (SUSHI, COGNAC-DPD)
Interface, phase separation simulation (SUSHI, COGNAC-DPD) -
 Rheology simulation (VSOP-DPD, PASTA, NAPLES)
Rheology simulation (VSOP-DPD, PASTA, NAPLES) -
 Multi-phase material, RVE, and Process simulation (MUFFIN, VSOP-PS)
Multi-phase material, RVE, and Process simulation (MUFFIN, VSOP-PS) -
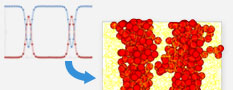 Multi-scale coupling function
Multi-scale coupling function -
 Data science function
Data science function -
 Platform Function
Platform Function
Crystal Structure and Molecular Modeling
Modeling tools for molecular dynamics calculations.
-
- All-atom modeling (1)
- Write a chemical formula directly on the screen for quick and easy 3D molecular structure creation. Create a polymer by combining the resulting monomer models. The system can construct a block copolymer or a random copolymer and control the tacticity. Importing files in various file formats including PDB, CIF, MOL and SMILES is supported. By specifying reaction points in an atomic element, the chemical reactions during the molecular dynamics calculation and the crosslinking can be defined. See also "Reaction modeling."
-
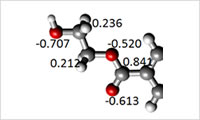
- All-atom modeling (2)
- For the force field parameters, AMBER, GAFF, DREIDING, OPLS-AA, L-OPLS, UFF, CLAY and PCFF are available for use. Direct Force Field (Team Force Field) is also available to obtain the force field parameters. What is "Direct Force Field"?
-
- All-atom modeling (3)
- By directly performing quantum chemical calculations including Gaussian, Firefly/ PC-GAMESS a point charge can be obtained.
-

- System modeling
- Construct a system by allocating the molecules that are generated by using an all-atom model or a coarse grained model. For a full atomic model, the system can be an amorphous structure or a crystal structure of the inorganic material. By merging these two systems, the organic-inorganic interface structure can be constructed. For a coarse grained model, the system can be constructed by creating filler using Kremer-Grest or DPD, or, by inserting filler into a polymer.
-
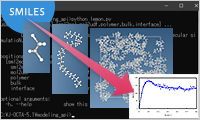
- MD modeling API
- J-OCTA supports automation of input file creation for MD calculation from any chemical structures expressed in SMILES notation. Creating both bulk and interface models is supported. Many calculations can be performed efficiently by using this function (high throughput calculations).
*...VSOP is a high-speed parallel molecular dynamics engine that is co-developed by the JSOL corporation and Japan Atomic Energy Agency (JAEA).
First-Principles Calculation (SIESTA)
First-Principles (FP) simulations make use the basic laws of quantum mechanics to give an insight of material properties such as electronic, mechanical, magnetic properties without making assumptions in empirical models or using fitting parameters. DFT (Density Functional Theory) calculation is one of the most popular methods in the FP simulation field. Current computational power enables to calculate properties of several hundreds to thousands of atoms with DFT.
Detail information of First-Principles calculation >>SIESTA is a registered trade-mark of SIMUNE.
SIMUNE Website >>
-
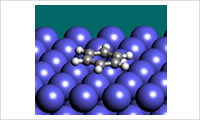
- SIESTA interfacial energy tool
- This tool gives an easy way to evaluate interaction energies between a surface and a molecule with SIESTA. It is possible to estimate surface adsorption energies from the interaction energy curve. Based on the interaction energies, force field parameters for molecular dynamics simulation can be estimated. Case Study: Example of Calculation with SIESTA
-
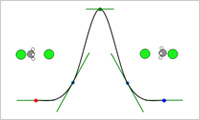
- SIESTA modeler
- This modeler gives an easy way to calculate stable structures, elastic moduli, reaction paths and so on using SIESTA. The activation energy, which is used as an input parameter of the reaction modeling, can be obtained from the reaction path analysis. Case Study: Cross-Linking of Epoxy Resin by the Monte Carlo method using activation energyDetail information of SIESTA modeler
Molecular dynamics simulation (COGNAC, VSOP, GENESIS)
Evaluate and estimate the static and dynamic properties of the material and molecules.
-
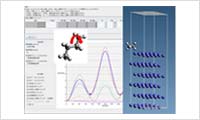
- Force field parameter adjustment
- This feature can adjust the bond potential in molecules and the LJ potential parameters for inorganic/organic interfaces, in accordance with the structure resulting from a quantum calculation (DFT or MO) and the potential energy. Automatic allocation of parameters is also available, and users can adjust the parameters with discussing comparisons of the given potential curve and the quantum calculation result.
-

- Coarse Grained modeling
- In the Coarse Grained modeling, an atomic assembly is treated as a unit (grained unit). Coarse grained models can calculate a large scale phenomenon that persists over a long period. The software possesses a function for estimating the coarse-grained potential based on the all-atom model. Case Study: Evaluation of Coarse Grained Potential
-
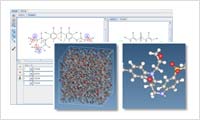
- Reaction modeling
- By specifying two molecules as reaction points in an atomic, users can setup various items such as parameters for the chemical reaction calculation using MD, a post-reaction structure, force field parameters, and the charge.
* VSOP is necessary for a reaction calculation using MD.
Case Study: Properties of Cross-Linked Phenolic Resins. Case Study of Epoxy 1 Case Study of Epoxy 2
-
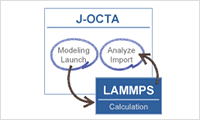
- External engine
- J-OCTA also possesses a function to convert the data to LAMMPS, GROMACS, HOOMD-blue and GENESIS, and can run these molecular dynamics engines directly on J-OCTA. What is LAMMPS?What is GENESIS?
-
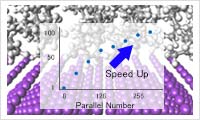
- Parallel molecular dynamics engine: VSOP
- VSOP (Parallel molecular dynamics engine) improves the calculation speed dramatically and enables large calculations that are not possible without parallelization.
-

- Post analysis
- In addition to the simulation function implemented in OCTA engine, J-OCTA can simulate the free volume, the primitive path, and the spherical model filling in a specified region. It is possible to export a density distribution for FEM simulation, in the format of a tetra mesh, a voxel mesh, or an STL.
-
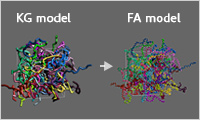
- Amorphous structure generation function using reverse mapping
- An amorphous structure of a full-atom model can be obtained at high speed by replacing the time-consuming relaxation calculations of the full-atom model with a Kremer-Grest model. The Kremer-Grest model is internally created from the full-atom model, a relaxation calculation is performed, and finally, the relaxed KG model is reverse-mapped to the full-atom model.
-

- GENESIS Modeler for Life Science
- Molecular dynamics calculations for biomolecular modeling and drug discovery will be supported. We plan to add useful functions such as protein-ligand docking structure search and free energy evaluation, and biomembrane modeling.
Review article : Drug discovery and formulation
*...VSOP is a high-speed parallel molecular dynamics engine that is co-developed by the JSOL corporation and Japan Atomic Energy Agency (JAEA).
Interface, phase separation simulation (SUSHI, COGNAC-DPD)
Estimate the phase separation structure and the interface geometry of materials with various molecular structure and block copolymers by using the mean field method (SUSHI) and the dissipative particle dynamics method (COGNAC-DPD).
-
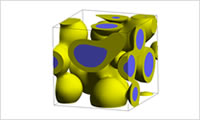
- Estimate calculation of the interaction parameters
- Estimate the interaction parameter (χ parameter). (1) Estimate the SP value (solubility parameter) by using the group contribution method, QSPR, or molecular dynamics to obtain the χ parameter. (2) Obtain the χ parameter based on the contact energy between the Coarse Grained segments by using force field calculations or fragment molecular orbital calculations, and the coordination number. (3) Back calculate the χ parameter based on the experimental phase diagram. Case study: Estimation of χ Parameters from Phase Diagrams
-
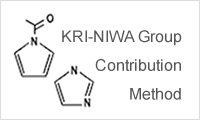
- KRI-NIWA method
- The KRI-NIWA method is a new group contribution method developed by KRI, Inc. that can estimate SP values more precisely than the traditional group contribution method. Estimate χ parameters based on the estimated SP values.
-
- χ parameter estimation using Monte Carlo method (FMO)(FCEWS)
- Interaction parameters (χ parameters) are estimated based on results of fragment molecular orbital calculations (FMO) using ABINIT-MP. Since the parameters are determined based on electronic state, general purpose calculations are possible even for molecular species that are difficult to evaluate in classical force field, for example, in the case of charge transfer being essential. FCEWS is a system developed by Mochizuki Lab., in Rikkyo University.
Detail of FCEWS
-

- Generate a mesh from the phase separation structure
- Convert the phase separation structure to an FEM mesh. Generate a mesh along the interface and export the volume fraction distribution into a finer mesh. (For the latter function, please refer to the description for MUFFIN as well.)
-

- Initial concentration distribution
- By combining multiple geometries (box, sphere, and cylinder), a concentration distribution or an obstacle domain can be created.
Rheology simulation (VSOP-DPD, PASTA, NAPLES)
Estimate the rheology property of the melted polymer or of the polymer considering the effects of the molar weight distribution and branch structure by using the slip link model (PASTA).
-

- Entanglement polymer model
- Build models considering the branch structure and crosslinking of the polymer and the molar weight distribution by using parameters such as the entanglement molecular weight. Shear and elongation flow are possible flow fields.
-

- Entanglement polymer model calculation with DPD
- The DPD calculation function implemented in VSOP can consider the entanglement effect of the polymer chain with slip spring. It allows users to evaluate material properties such as viscoelasticity.Case study
-
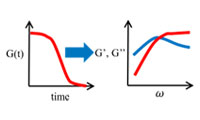
- Evaluation of viscoelasticity using simulation results
- The viscoelasticity (master curve) is calculated using the relaxation modulus obtained from the molecular simulation. It is also possible to obtain the Prony series. i-Rheo GT developed by Dr. M. Tassieri is partially used.Case study
Multi-phase material, RVE, and Process simulation (MUFFIN, VSOP-PS)
Finite element simulations are performed using phase-separated structures obtained by mean-field methods and other methods. It is also possible to simulate complex flow fields such as particulate dispersion systems using the particle method.
RVE analysis function in J-OCTA
-

- Import the phase separation structure
- Import the phase separation structure obtained by the mean field method or DPD as an elastic modulus distribution on the FEM mesh. Estimate the stress distribution and the energy distribution on the interface and the mean elastic modulus that are obtained by the elastic simulation using MUFFIN.
-

- Meshing
- Generate an FEM mesh along the interface by allocating simple spheres inside the rectangular domain. The result can be used to simulate the virtual phase separation structure and the composite materials.
-

- Multi-layer fluid simulation
- Build models to perform a simulation with a micro-fluid that contains multiple polymers and/or electrolytes. These models are applicable to the calculation of thin film formation with phase separation (e.g., solvent evaporation) and other related phenomena.
-
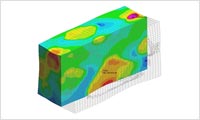
- External engine
- Ansys LS-DYNA link : The phase separation structure in voxel mesh which is output from SUSHI (SCF), DPD or CGMD can be used to run Ansys LS-DYNA structural analysis (uniaxial tensile, biaxial tensile, and shear deformation) and thermal analysis. The material can be elastic, elastoplastic, hyperelastic, or viscoelastic. * This function will be released in next version. Refer Ansys LS-DYNARefer the interface to Ansys LS-DYNA
-
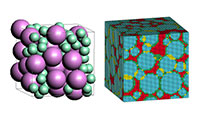
- Representative volume element (RVE) for composite materials
- COGNAC can create a structure (Representative Volume Element(RVE)) of a rectangle region which is highly filled (over 50 vol%) with particles, fibers, and discoid shaped fillers. The resulting structure can be transferred to Digimat-FE and be output as an STL file and a voxel mesh. These files can be used for FEM simulation for composite materials. Refer Digimat-FERefer RVE modeler
-
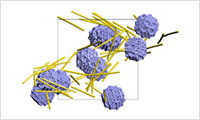
- Process Simulation (particulate dispersion systems, droplets, coatings)
- Fluid dynamics analysis can be performed using the MPS (particle method) functionality included in VSOP-PS. It targets complex fluid phenomena such as flow fields in particulate dispersion systems and evaporation phenomena from droplets and coating films (under development).Case StudyVSOP-PS(Particle Simulation)
Multi-scale coupling function
Determine the molecular construction of polymers by using a large scale simulation result. The large scale result employs multi-scale approaches and enables the generation of a relaxation molecular construction that is complicated when general molecular dynamics is used.
-
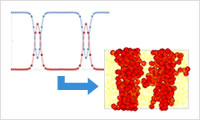
- Zooming
- Create the molecular dynamics structure based on the volume fraction distribution (phase separation structure) of each component obtained with the mean field method (SUSHI). This function is especially useful to create the molecular construction around the interface.
-

- Reverse mapping function
- Create the all-atom molecular dynamics structure by using the molecular construction obtained by the coarse-grained molecular dynamics. The resulting structure can be used to create the initial structure of the all-atom model and the evaluation of the coarse grained model (to be compared with the experiment).
Data science function
Machine Learning functions for Materials & Process informatics. Prediction of physical properties from the molecular or crystal structure, and inverse analysis is possible. Other useful functions are implemented.
Review article : Materials & Process Informatics
Property estimation function using machine learning "MI-Suite (Materials Informatics Suite)"
-

- Quantitative Structure Property Relationship(QSPR)
- This function can estimate ranges of material properties of polymers from input molecular constructions. This function outputs a list of material properties including the density, thermal expansion coefficient, glass transition temperature, Poisson's ratio, and dielectric constant. This is a powerful tool for supporting the molecular design of an organic polymer. Case Study: Estimation of Property Values Using QSPR
-
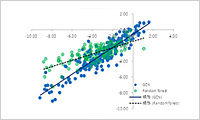
- Machine Learning based QSPR(ML-QSPR)
- Machine learning using GCN, CGCN, XGBoost, etc. is used to learn the relationship between various parameters such as molecular structures and processes and their physical properties. The system then predicts the physical property values under unknown molecular structures and conditions. It also includes a transfer learning function. Molecular structures in various formats such as SMILES are supported. We also provide learned libraries and a database of physical properties.MD modeling APICase study
-
- mol-infer (Inverse QSPR)
- J-OCTA interfaces with mol-infer, which is developed by Nagamochi Laboratory at Kyoto University in Japan. The inverse analysis, i.e., a method to predict molecular structure from physical properties is available. The inverse operation is solved by Mixed Integer Linear Programming (MILP). Detail information >>
-
- Analysis & public DB connection
- The latest functions related to data science, such as the ability to obtain physical property data by connecting to public DBs such as Materials Project and PubChem, and the ability to extract common parts from multiple molecular structures, are constantly being updated.
-
- Molecular descriptor calculation function
- This function calculates and outputs molecular descriptors required for machine learning, using SMILES and MOL formats as input.
· 2D descriptors
· 3D descriptors
· Fingerprints
· Graph structures
-
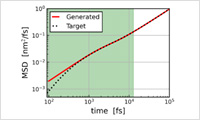
- MD-GAN
- This function predicts the motion of molecules over a long period of time by machine learning using the results of short-time molecular dynamics (MD) calculations. This method was developed by Yasuoka Laboratory at Keio University in Japan. For example, it is possible to predict diffusion coefficients quickly, whereas long-time MD calculations are usually required. Detail information >> Case study
Function Details
- +QSPR
- Estimated material
- - Polymer materials composed of carbon, hydrogen, oxygen, nitrogen, silicon, sulfur, fluorine, chlorine, and bromine
- - van der Waals volume
- - Molar volume
- - Density
- - Coefficient of volume thermal expansion, Coefficient of linear thermal expansion
- - Heat capacity at constant pressure
- - Amount of change in the heat capacity near the glass transition temperature
- - Cohesive energy, Solubility parameter, χ parameter
- - Glass transition temperature
- - Surface tension, Interfacial tension
- - Refractive index, Molar refraction
- - Dielectric constant, Molar polarization, Effective dipole moment, Dissipation factor
- - Molar diamagnetic susceptibility
- - Poisson's ratio, Bulk modulus, Shear modulus, Young’s modulus
- - Entanglement molecular weight
- - Stress of brittle fracture, Yield stress
- - Characteristic ratio
- - Zero shear viscosity
- - Thermal conductivity, Thermal diffusivity
- - Permeability (oxygen, nitrogen, carbon dioxide)
- - Temperature of Half-decomposition
- - Molar Thermal Decomposition Function
- +ML-QSPR
- Prepared data set
- - Density
- - Glass transition temperature
- - Characteristic ratio
- - Dipole moment
- - HOMO/LUMO
- - Further datasets will be added.
- - Your physical data is available to be learned.
Platform Function
-

- Link and run engines
- Direct link to OCTA engine as well as the external engines allows users to perform a local run (on Windows) or a remote run (on Linux) . Interface access to the job management system is also available. A parametric calculation for given simulation conditions is possible.
-

- 3D drawing
- 3D drawing feature supports a small-scale molecular drawing for editing a model as well as a large-scale model drawing. It is possible to draw a model which contains millions or tens of millions of particles.
-
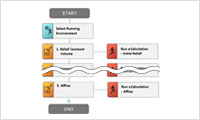
- Scenario
- Simulation procedure can be saved as a scenario which is created in an easy-to-understand flowchart style. By registering a scenario, users can repeat a common simulation procedure with different materials. Scenario is useful to share a procedure in your team as well.
System Requirements
J-OCTA
| OS |
|
|---|---|
| CPU | Multi-core CPU (recommended) |
| Memory | 16 GB or more (recommended) / 8 GB (required) |
| Hard disk |
|
| Display resolution | 1024 X 768 or more (recommended) |
| Graphics card | OpenGL-compatible graphics card (nVidia is recommended) |
| Display colors | 65536 colors or more (recommended) |
Analysis Engine
| OS |
|
|---|---|
| CPU | Multi-core CPU (recommended) |
| Memory | 16 GB or more (recommended) |
| Hard disk |
|