SIESTA Modeler
Currently, SIESTA is available as free First-Principles (ab initio) calculation software (engine) and is used in many countries and regions around the world.
SIESTA is a registered trade-mark of SIMUNE.
SIMUNE Website >>
Detail information of First-Principles calculation >>
SIESTA's Strengths are below.
✓ SIESTA uses atomic(local)-orbital basis, it has advantages over plane-wave basis software for surfaces, molecules, and two-dimensional materials. Especially in the vacuum region, plane-wave basis requires a lot of computational resources, whereas SIESTA does not.
✓ SIESTA uses atomic(local)-orbital basis, it has advantages over plane-wave basis software for surfaces, molecules, and two-dimensional materials. Especially in the vacuum region, plane-wave basis requires a lot of computational resources, whereas SIESTA does not.
✓ To compute the van der Waals forces, SIESTA implements a variety of van der Waals functionals, rather than the corrections common in other software.
However, even though it is free software, it is not easy to use for people who are not experts in first-principles calculations (density functional theory: DFT), because it is necessary to do everything from preparation of input data to analysis of results.
Therefore, J-OCTA has prepared a dedicated GUI, the SIESTA Modeler, so that even non-specialists can easily use the SIESTA code. This makes it easy for non-specialists to perform first-principles calculations using SIESTA and evaluate various physical quantities and properties. SIESTA modeler is developed by SIMUNE (https://www.simuneatomistics.com/).
The figure below shows the SIESTA modeler screen, which is very simple but allows various calculations to be performed. For example, you can select a project type of single point calculation (where the atomic configuration is unchanged and only the energy is calculated) or optimization calculation (where the atomic configuration is moved), and then configure the parameters for each calculation in the project settings. The setup and calculations can be performed by clicking from left to right.
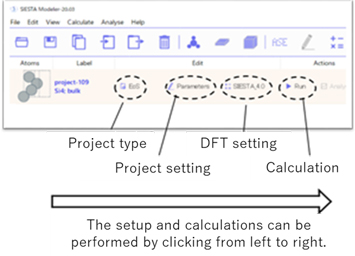 Fig 1. Features of the SIESTA modeler
Fig 1. Features of the SIESTA modeler
In DFT calculations using SIESTA, the "numerical basis functions" and "pseudopotentials" set for each element have a significant impact on calculation accuracy and convergence. The SIESTA modeler contains data tuned by SIMUNE, which enables highly accurate and fast calculations.
 Fig. The SIESTA modeler sets the appropriate data for each element.
Fig. The SIESTA modeler sets the appropriate data for each element.
With the SIESTA modeler, several physical quantities and properties can be easily obtained without being an expert. For example, even when evaluating familiar physical properties such as modulus of elasticity, technique is sometimes required, but the SIESTA modeler allows you to take advantage of past expertise. The NEB (Nudged Elastic Band) calculation function can be used to analyze the activation energy of a chemical reaction, the EOS (Equation of State) calculation function can be used to calculate the bulk modulus of a solid, and phonon analysis can provide information on the lattice specific heat and modulus. Ab Initio MD (AIMD) calculations can also be used to analyze thermal expansions and reactions.
The following figure is an example of the actual SIESTA modeler screen.
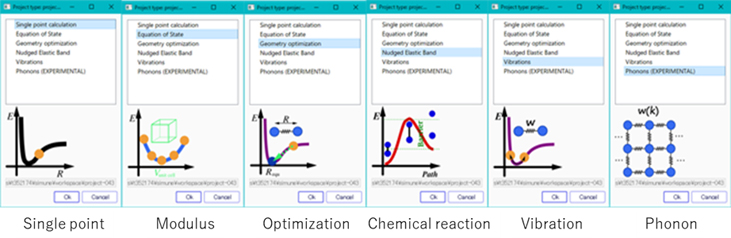 Fig. Examples of objects that can be calculated with SIESTA modeler
Fig. Examples of objects that can be calculated with SIESTA modeler
Molecular dynamics (MD) engines such as COGNAC, VSOP, GROMACS, LAMMPS, and GENESIS included in J-OCTA may be used to perform "organic/inorganic interface" calculations. The calculation targets include electrode interfaces, adhesive and filler surface design, etc. Please refer to the J-OCTA case study list for details. The problem in such cases is the force field parameters. It is difficult to find force field parameters that describe the interactions between the atoms that make up inorganic materials and organic molecules, respectively.
Using SIESTA, we can evaluate energy curves by placing organic molecules on an inorganic surface (exhaustive and allowing for rotation of the molecules) and then performing DFT calculations at varying distances from the surface. Furthermore, J-OCTA has the ability to identify force field parameters that reproduce the obtained energy curves, allowing MD calculations for organic/inorganic interfaces.
As mentioned earlier, the strength of SIESTA is demonstrated in such calculations: SIESTA and other DFT calculations of this type are subject to periodic boundary conditions, which means that the same structure is repeated in each direction of the simulation box. This means that the same molecule will be right next to the organic molecule to be adsorbed, and the surface area must be as large as possible (to avoid interference between molecules). Also, the thickness of the inorganic crystal (slab structure) must be a certain amount, which inevitably increases the number of atoms. SIESTA, which uses a numerical localized basis, consumes less memory than software using a plane-wave basis, and furthermore has an advantage in calculating inhomogeneous structures, such as those containing vacuum regions. In addition, SIESTA includes features suitable for van der Waals force evaluations, making it suitable for such evaluations.
The figure below shows the energy curves of propane, butane, pentane, and hexane adsorbed on a copper surface, where the dots are the SIESTA (DFT) results and the solid line is data using force field parameters identified from the DFT results.
The solid line is the data using the force field parameters identified from the DFT results. Since the force field parameters common to all molecules were obtained, the solid line and the dots are slightly different in some places, but this can be adjusted.
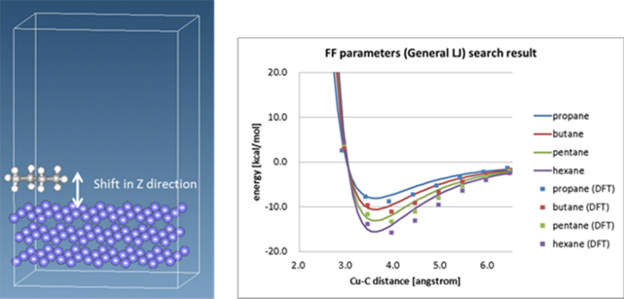 Interfacial energy curves obtained with SIESTA and fitted Force Field
Interfacial energy curves obtained with SIESTA and fitted Force Field